
Long reads are getting longer – again!
AGI leverages again its expertise in handling large DNA fragments
Over the past few weeks, AGI’s wet lab team has been working on the development of larger PacBio HiFi libraries with the goal of obtaining genome assemblies featuring higher contiguity and completeness.
The effort began with optimizing genomic DNA shearing conditions on the Diagenode Megaruptor 3, aimed at having most of the DNA between 15 and 40 kb. Optimal shearing conditions coupled with early access to a new Sage Science Pippin HT Range + T mode (software version v1.14-CD17), allowed to accurately size-select DNA starting from 15 to 20 kb. Agilent’s Femto Pulse was used to size the DNA at all stages of sample preparation.
Below, the results on a plant sample. Genomic DNA was sheared at two conditions (black trace, speed 30; blue, speed 28 and 29), and a whole-genome shotgun library was prepared with PacBio SMRTbell Prep Kit 3.0. The library was size selected at a minimum size of 15 kb (red trace) and the recovery rate was 20.3%. The library was sequenced in three SMRT cells 8M on a Sequel IIe.

The sequencing yielded an average of 28.7 Gb and 1.26 million reads per cell, a QV score of 29, and an average HiFi read length of 22.6 kb. The histogram insert illustrates the HiFi read length distribution.
Three other plant DNA samples (K, M, and A) were sheared to a mode of 22-25 kb and size-selected with the Pippin HT at different values between 17 and 20 kb. The HiFi read length distribution below shows: i) the efficiency of the gel-based size selection at removing the DNA below the threshold; ii) the accuracy and consistency of the selection at the cutoff value; iii) the long tail with many molecules at > 30 kb, important when developing de novo assemblies of new species. Since each sample sheared slightly differently, we selected each DNA at two different thresholds. The effect of the cutoff value is clearly noticeable, resulting in libraries of 23-24 kb.

We applied the same protocol to additional samples, varying the shearing and size selection conditions. We tested 10 different libraries belonging to 4 plant species, sequencing a total of 27 cells. The scatterplot displays the distribution of HiFi mean read length (x axis) and yield (y axis), respectively. Without considering two cells that underperformed due to loading issues, the average yield per cell was above 28 Gb, with mean read lengths above 19 kb in 8 of the 10 samples (blue dots).
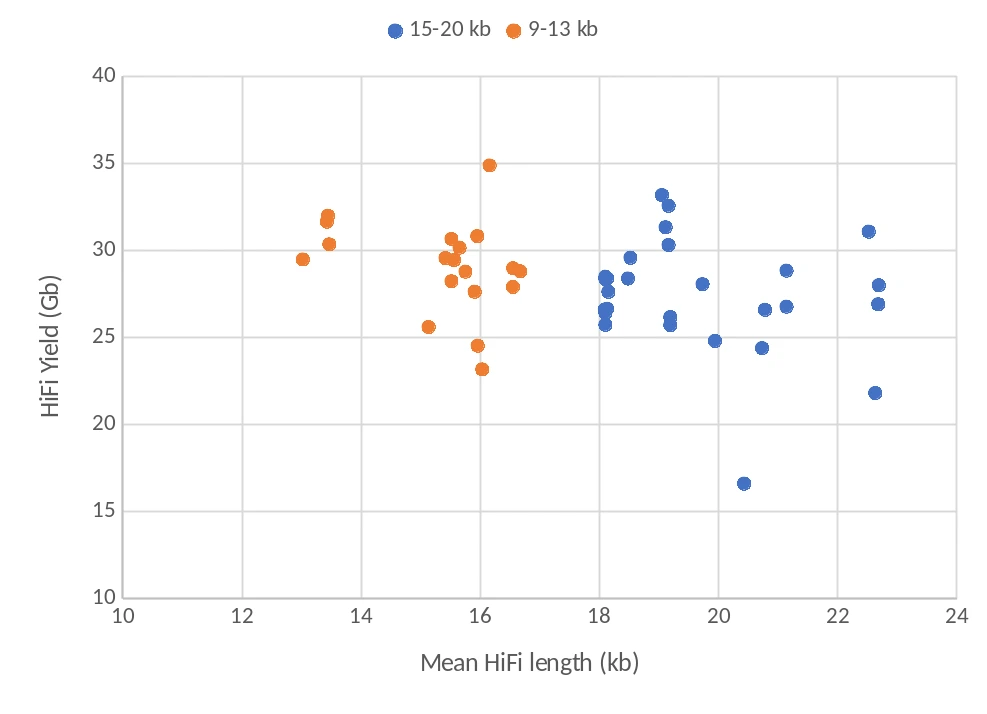
Compared to a subset of previous libraries (orange dots, selected with Pippin HT software v1.13, 6 samples, 19 SMRT cells, 29.1 Gb mean yield per cell, mean read length 15.3 kb), the increase in read length coming from the updated Pippin HT size selection mode was noticeable and consistent. Two samples were processed in both conditions, and each saw a 6 kb increase in read length when the DNA was selected with the R+T mode.
To test whether longer reads lead to more contiguous assemblies, we assembled the data of the sample presented above, for which we had both conventional (16 kb mean read length) and large (22 kb) libraries. To assess exclusively the effect of read length on assembly contiguity, we normalized the datasets (i.e. compared assemblies obtained from the same amount of bases but different read lengths).
In the table below, both at low (7.8×) and intermediate coverage (12×), the number of contigs decreases by almost 30%, with total assembly size being the same. Importantly, assembly Nx statistics also improve when assembling with longer reads. The improvement ranges from 25% to 40% at low coverage and decreases slightly (2-15%) at higher coverage. This lower increase may be due to the fact that the additional coverage came from shorter reads (overall mean size 19.9 kb). Nevertheless, just with less than 8× coverage, a 11 Gb, tetraploid genome was assembled at 84% completeness in 12,152 contigs, with N50 of 1.58 Mb and the longest contig was 12.4 Mb.
| Low coverage, conventional length | Low coverage, longer reads | Intermediate coverage, conventional length | Intermediate coverage, longer reads | |
| Number of reads | 5,162,259 | 3,798,962 | 8,143,559 | 6,780,262 |
| Total bases (Gb) | 85.65 | 85.96 | 133.33 | 133.64 |
| Genome coverage (×) | 7.8 | 7.8 | 12.0 | 12.0 |
| Mean read length (kb) | 16.3 | 22.6 | 16.3 | 19.9 |
| Assembly length (Gb) | 9.32 | 9.28 | 9.44 | 9.42 |
| Number of contigs | 15,713 | 12,152 | 5,318 | 4,186 |
| Mean contig length (Mb) | 0.59 | 0.76 | 1.78 | 2.25 |
| Longest contig (Mb) | 8.31 | 12.43 | 78.27 | 57.60 |
| N50 (Mb) | 1.14 | 1.59 | 7.15 | 8.20 |
| L50 (#) | 2,406 | 1,702 | 349 | 303 |
| N70 (Mb) | 0.72 | 0.97 | 4.14 | 4.52 |
| L70 (#) | 4,474 | 3,195 | 696 | 612 |
| N90 (Mb) | 0.32 | 0.40 | 1.60 | 1.64 |
| L90 (#) | 8,228 | 6,092 | 1,413 | 1,284 |